Projects
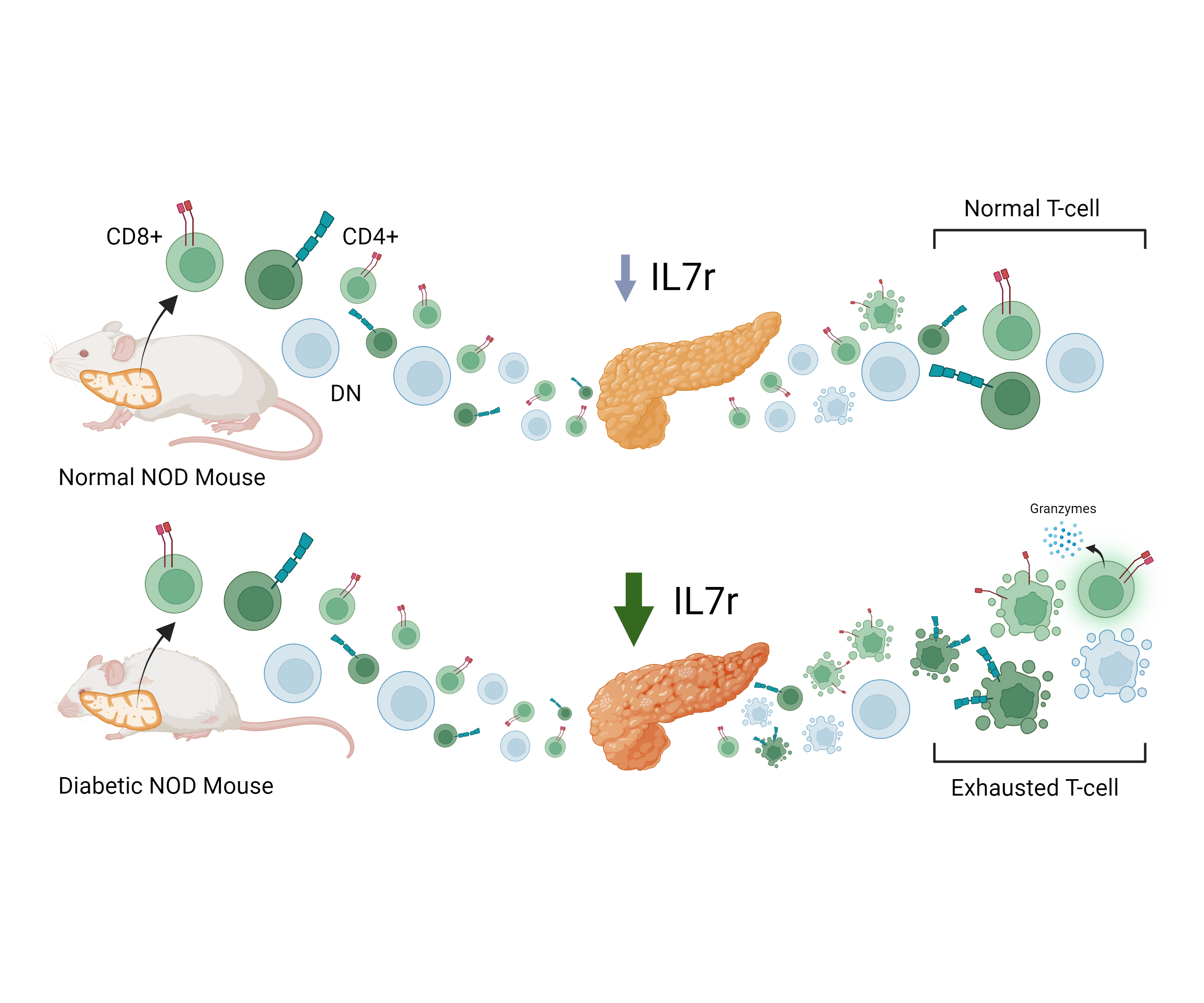
Environmental effects and dysregulation of T-cells in inflammatory diabetes
Type-1 Diabetes is an inflammatory autoimmune disease that develops during childhood where immune dysregulation leads to Beta-cell destruction and a loss of the bodies ability to regulate insulin production. This disease affects millions worldwide and is curiosly more common in highly modernized western countries. One of the most well known examples of this is the 6x greater risk of disease in finnish juveniles compared to russian juveniles. Currently, this is difficult to study due to high heterogenity in human populations. Our lab aims to develop both computational and in-vivo mouse models to better understand how environmental factors affect disease development.
We have recently published one of the first single cell RNA-seq studies in the translational NOD mouse model and found 1) High proportions of Double Negative T cells 2) Clonal specific LAG3+ CD8+ T cells and 3) Immune metabolic dysregulation of pancreatic infiltrating T cells. To investigate these results further, our lab combines novel sequencing methods and in-vitro experimentation.
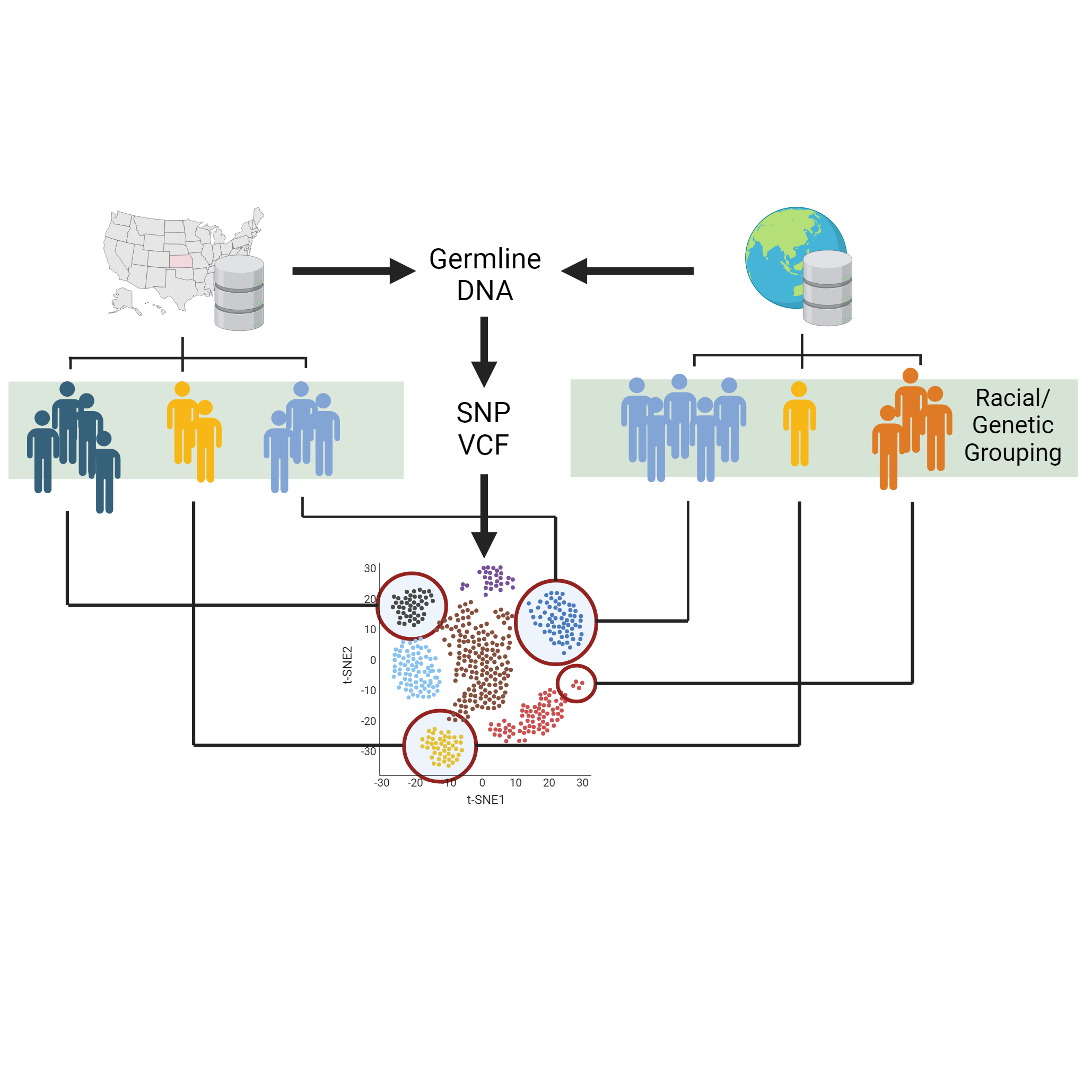
Exploring environmental effects on cancer progression through international sequencing data
Racial and genetic bias towards the development of cancer is a major factor that disproportionally impacts at-risk populations in this country. Black American populations are at higher risk of developing and dying from prostate and colorectal cancer, while Asian American populations are the only which have seen increases in breast cancer mortality in the last decade. Interestingly, native populations of genetically similar ethnic groups do not experience the same incidence or mortality of disease with their genetic matches in the US. This perplexing nature of unequal bias in cancer development has gone long unresearched by the wider scientific community due to geographical barriers in patient groups, but has become more possible now thanks to the wide abundance of publically available sequencing data. Using this data, we can conduct international investigation into patients suffering from similar cancer and observe how the combined effect of genotype and environment result in the different pathology among individuals. Through this research we hope to identify biomarkers or risk factors unique to individual ethnic groups to improve health equity and cancer survival.
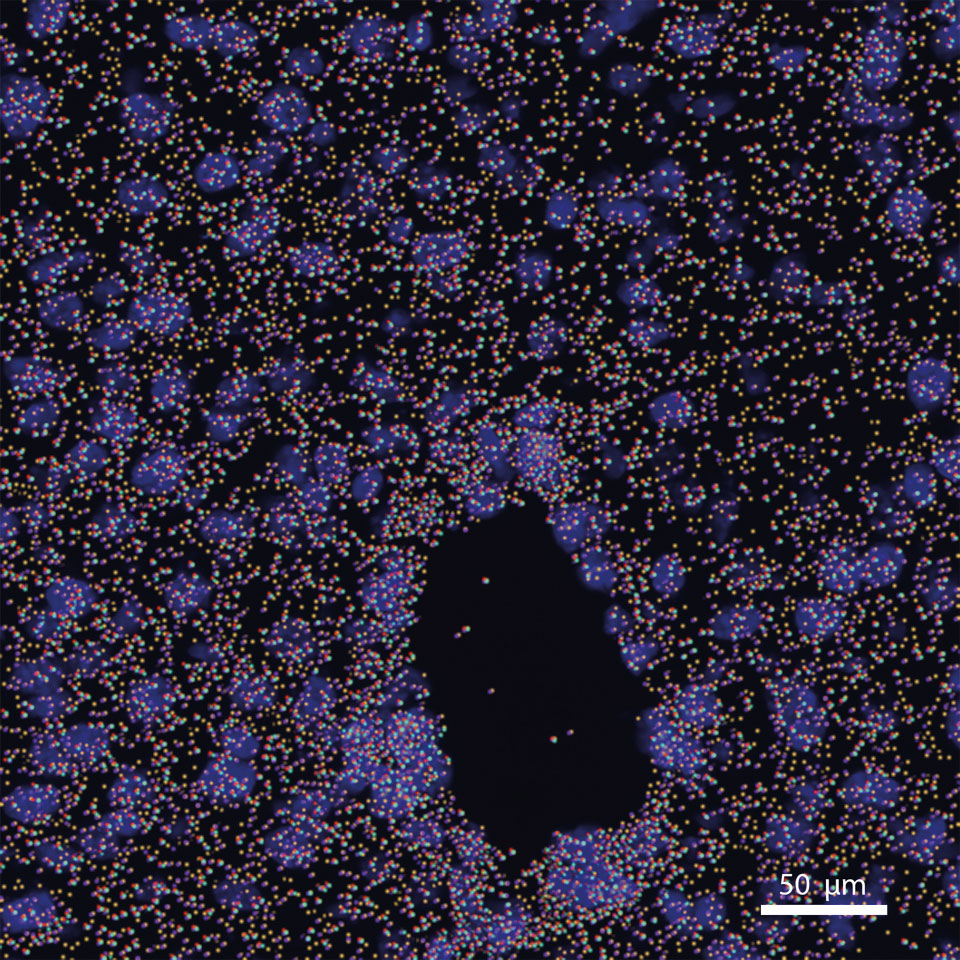
Computational methods for the analysis of spatial and single-cell sequencing of the tumor microenvironment
Single cell and spatial omics technologies offer a new way to understand the biology of tumor development and immune cell interaction. Due the immaturity of this technology the methods to generate this data is much more developed than the methods for analysis. Topics that still need development are clustering algorithms and cell type identification, low sequencing depth and sampling error, and the recognition of cell-cell interactions/signaling events. In our lab we use computer vision algorithms which include vision transformers, resnets, and unsupervised learning to address these opportunities. A focus of our research is establishing a universal method of gene expression modeling for the different technologies of spatial transcriptomics that will standardize resolution to the single-cell level.
Packages
Coming soon…
Funding Sources
